
|
||||||||||||||||||||||||||||
| Table 1: Bonding energies in different units. [7] |
The Fischer-Tropsch process is a gas to liquid (GTL) polymerization technique that turns a carbon source into hydrocarbons chains through the hydrogenation of carbon monoxide by means of a metal catalyst. The feedstock is typically coal or natural gas, though more exotic (and carbon neutral) possibilities such as removing CO2 from the ocean or the atmosphere have been considered. [1,2] Biomass could serve as feedstock but is not cost competitive for transport fuels. [3,4] In terms of economies of scale, current plants can produce tens of thousands of barrels per day with the largest plant (Shell's Pearl GTL in Qatar) producing 140,000 barrels per day. [5]
The carbon source is converted to syngas, a combination of carbon monoxide (CO) and hydrogen (H2) gas, through a process of gasification (C + H2O → CO + H2) where a controlled flow of steam and oxygen is maintained through the source at high temperature and pressure (1200 - 1400°C and 3 MPa ~ 30 atm) without enough oxygen for complete combustion. [6] Different feedstocks and syngas production mechanisms produce different H2/CO ratios; coal gasification produces ratios close to unity whereas steam reforming of natural gas produces a ratio of 3/1. [4] The syngas production process consumes 60- 70% of the capital and running cost of the plant, so the proper ratio for the Fischer-Tropsch reaction must be maintained to maximize syngas conversion efficiency. [6] This ratio is controlled by an exothermic water-gas shift reaction (CO + H2O → CO2 + H2) to increase H2/CO.
The resulting syngas is passed through a metal catalyst which causes polymerization into hydrocarbon chains via the reaction (n CO + (2n+1) H2 → Cn H 2n+2 + n H2O); since the Fischer-Tropsch reaction is exothermic, excess heat needs to be removed to avoid deactivating the catalyst and increasing methane production. [6] We can consider the bonding energy associated with adding a single CH2 to the hydrocarbon chain by the reaction: (CO + 2 H2 → CH2 + H2O). Including the extra bond for the carbon chain, we have:
Taking the values for the bond energies in Table 1, the energy change is EFT = -1.55 eV. [7]
|
|
| Fig. 1: Weight fraction sum for gasoline and diesel. |
Assuming the chain growth probability is independent of the chain length, the polymerization is described by the Anderson-Schulz-Flory distribution Pn = αn-1(1-α) where Pn is the probability of producing a hydrocarbon of length n (mole fraction). [8] The distribution depends on a single parameter: α, the probability a chain will grow rather than desorb from the catalyst. αn-1 is the probability of adding n-1 carbons, and (1-α) is the probability of not adding a carbon and therefore terminating chain growth. The expected chain length is given by <n> = ∑ n Pn = 1/(1-α) such that the weight fraction Wn = Pn (n/<n>) for each n is given by [5]
The maximum probability Pn with respect to α (dPn/dα = 0) occurs at α = (n-1)/(n+1) and is given by Pn = 2/(n+1) × (n-1/n+1)n-1. By summing the weight fractions Σn Wn, we see that the maximum yield is about 45% by weight for gasoline (n=5-11) occurring at α = 0.75 and about 25% for diesel (n=12-18) occurring at α = 0.87 (see Fig. 1). [9] The overall yield is then improved by combining shorter chains (propane n=3 and butane n=4) produced in the Fischer-Tropsch synthesis; this oligomerization can be facilitated with zeolite, a microporous mineral consisting of aluminum, silicon, and oxygen, as a catalyst. [9]
The chain growth probability (α) can be tuned by the temperature, syngas composition, pressure, catalyst choice, and presence of promoters. [6] A higher CO partial pressure results in increased surface coverage and therefore larger chain growth probability; a higher H2 partial pressure leads to chain termination and therefore smaller chain growth probability. [6] Independent of feedstock or catalyst, a high reaction temperature leads to faster hydrogenation (significant methane CH4 formation and less surface CH2) and therefore favors a shorter average chain length. [3,6,9] High temperature additionally results in elemental carbon deposition via the Boudouard reaction (2CO → CO2 + C) that deactivates the catalyst. [6] In terms of bonding energy, we have:
where the elemental carbon bond is included. The reaction energy is then EB = -1.42 eV, which is less (0.13 eV = 1574°C) than the Fischer-Tropsch reaction. [7] It follows that an efficient cooling system is necessary to produce transport fuels and maintain an activated catalyst.
A metallic catalyst serves to polymerize the syngas into hydrocarbon chains via the reaction n CO + (2n+1) H2 → CnH2n+2 + n H2O. The metal catalyst consists of iron, cobalt, or ruthenium. [8] While nickel is a highly active, it results in a large fraction of methane and is therefore unsuited for transport fuels. [6,9] Other group VIII metals have lower activity and less control for selecting longer chain hydrocarbons. [8] These transition metals all have partially filled d orbitals and are ferromagnetic. The catalysts are sensitive to sulfur contamination in the syngas and carbon deposition. [3,4,6] Iron is more susceptible to carbon, and cobalt is more susceptible to sulur. [3]
Iron catalysts operate at 300 - 350°C with H2:CO = 1.7:1. [4,6] Iron catalysts undergo the water-gas shift reaction more readily than cobalt making it the better choice for CO-rich syngas such as coal or biomass (and similarly a poor choice for H-rich syngas from natural gas). [3,5] Other ferromagnetic phases of iron including iron nitride, iron carbide, and iron carbonitride also show high Fischer-Tropsch activity. [8] While iron nitrides tend to oxidize at a slower rate, they produce oxygenated molecules and shorter chains. [8] The chain growth probability (α) can be increased with the addition of alkali promoters such as K2O to enhance CO dissociation. [3,6,9]
Cobalt catalysts operate at a lower temperature 200 - 240°C with H2:CO = 2.15:1. [4,6] Cobalt is about 1,000 times more expensive than iron, so it is necessary to maximize the catalyst surface area to minimize cost. [6] This is done by dispersing cobalt onto aluminium oxide, silicon dioxide, or titanium dioxide supports with ratios of 10-30% of cobalt to support. [6] Unlike iron, cobalt catalysts aren't strongly influenced by promoters; small concentrations of ruthenium, rhenium, and platinum have been shown to increase activity, but it is unclear whether the chain growth probability, and therefore hydrocarbon selectivity, is affected. [6] Cobalt catalysts used for the production of diesel fuel from natural gas are designed to maximize paraffin wax production. [3] The wax then undergoes hydrocracking, where long hydrocarbon chains are broken to shorter chains through heating and hydrogen saturation, to increase the diesel yield to over 73%. [9]
Ruthenium can also be used as a catalyst, but it is 50,000 times more expensive than iron. [6,9] Ruthenium has the advantage of working at a lower temperature (but higher pressure) than the other metals and producing the highest molecular weight hydrocarbons without the addition of any promoters. [3] Since it functions as a pure metal, this additionally makes ruthenium the ideal catalyst to study the mechanism behind the Fischer-Tropsch synthesis.
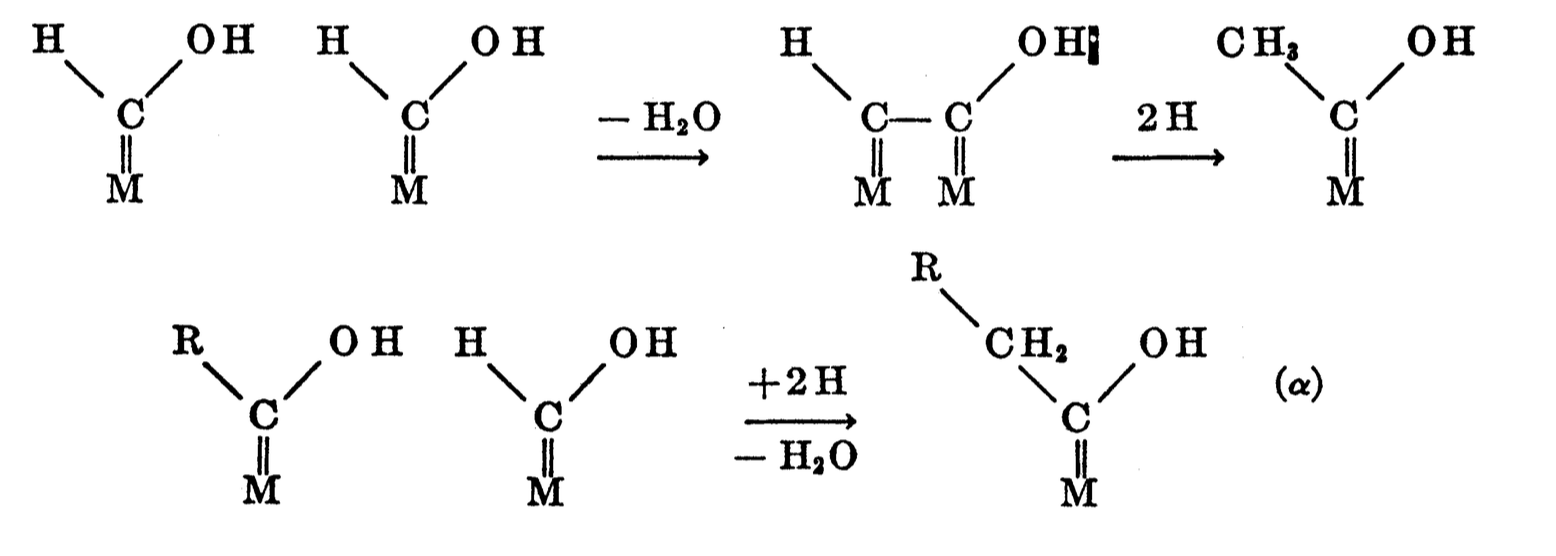 |
| Fig. 2: Hydrogenation of surface carbonyl. [8] (Courtesy of the U.S. Department of the Interior) |
The Fischer-Tropsch reaction can be visualized as a repeated chain growth process (CO + 2 H2 → CH2 + H2O) where hydrogen is added, the C-O bond is broken, and a new C-C bond is created. [3] The CO adsorption onto the catalyst surface can either be dissociative (C + O separately attached to the surface) or nondissociative (CO attaches to the surface). The microscopic question is then whether (1) hydrogen is first added to adsorbed CO such that there are intermediate, oxygenated products or (2) the CO bond is split first resulting in dissociative adsorption and intermediate hydrocarbon chains. [3]
Fischer originally proposed a ''carbide mechanism'' where CO and H2 are dissociatively adsorbed onto the catalyst surface, the carbon and oxygen are hydrogenated, the H2O desorbs, and the adsorbed CH2 continues the polymerization process. [10] The experimental signature of this process is carbide formation, which was observed in iron but was not found in other catalysts. [3] Stroch, Golumbic, and Anderson suggested surface carbonyls (carbon bonded to the surface and doubly bonded to oxygen) form by nondissociative adsorption. Hydrogen is then added to both the carbon and oxygen. [3,8] As can be seen in Fig. 2, the carbon chain is formed by releasing a water molecule and hydrogenating the carbon atom without oxygen. The hydrogenated carbon then desorbs from the surface, and chain growth continues. Experimental support for the surface carbonyl mechanism is found in the spectroscopic analysis of CO hydrogenation under high vacuum on a ruthenium catalyst. [11] Spectroscopic peaks for the CHO intermediates with (1) only carbon bonded to the surface (and doubly bonded to oxygen) and (2) carbon doubly bonded to the surface and oxygen bonded to the surface were both observed.
Density functional theory calculations studying the Fischer-Tropsch pathway on cobalt also argue against the carbide theory and start with nondissociative adsorption of CO. [10] This is followed by hydrogen(s) attaching to the carbon atom to weaken the carbon-oxygen bond and lower the CO dissociation energy barrier. Both the carbon and oxygen are bonded to the cobalt surface, and the product after dissociation is the hydrocarbon building block CH2. This reaction pathway was concluded from a comparison of the activation barriers and dissociation energies. The calculations showed an adsorbed CO dissociation barrier of 2.82 eV but an adsorption energy of 1.8 eV suggesting direct CO dissociation (i.e. without hydrogen) is unfavored. The addition of the first hydrogen (CO + H → CHO) had an activation barrier of 1.31 eV and addition of the second hydrogen (CHO + H → CH2O) only 0.45 eV. The calculations also showed the oxygen atom of CHO and CH2O bonds to the surface which reduces the barrier for breaking the carbon-oxygen bond to 1.0 eV (CHO → CH + O) and 0.85 eV (CH2O → CH2 + O). This is slightly different from the Stroch mechanism where the second hydrogen bonds with the oxygen; the calculations found a slightly higher activation energy of 0.81 eV.
While the numerical evidence points towards hydrogenation as the main reaction pathway, we need to study the experiments to determine if this is truly the case. The CHO is short-lived and exists only at high pressure so that is the first regime to search for experimental verification. [10]
© John Dodaro. The author grants permission to copy, distribute and display this work in unaltered form, with attribution to the author, for noncommercial purposes only. All other rights, including commercial rights, are reserved to the author.
[1] B. Klopfer, "Seawater to Jet Fuel," Physics 240, Stanford University, Fall 2012.
[2] D. Sleiter, "Synfuel Cycle Efficiency," Physics 240, Stanford University, Fall 2010.
[3] J. Schulz, "Short History and Present Trends of Fischer-Tropsch Synthesis," Appl. Catal. A 186, 3 (1999).
[4] P. L. Spath and D. C. Dayton, "Preliminary Screening - Technical and Economic Assessment of Synthetic Gas to Fuels and Chemicals with Emphasis on the Potential for Biomass-Derived Syngas," U.S. National Renewable Energy Laboratory, NREL/TP-510-34929, December 2003.
[5] C. Perego, R. Bortolo, and R. Zennaro, "Gas to Liquids Technologies For Natural Gas Reserves Valorization: The Eni Experience," Catal. Today 142, 9 (2009).
[6] Dry, M.E. "The Fischer-Tropsch Process: 1950-2000," Catal. Today 71, 227 (2002).
[7] R. T. Sanderson, Chemical Bonds and Bond Energy, 2nd Ed. (Academic Press, 1976).
[8] R. B. Anderson et al. "Physical Chemistry of the Fischer-Tropsch Synthesis," U.S. Bureau of Mines, Bulletin 580, 1959.
[10] M. E. Dry,"Practical and Theoretical Aspects of the Catalytic Fischer-Tropsch Process," Appl. Catal. A 138, 319 (1996).
[11] O. R. Inderwildi, S. J. Jenkins, and D. A. King, "Fischer-Tropsch Mechanism Revisted: Alternative Pathways for the Production of Higher Hydrocarbons from Synthesis Gas," J. Phys. Chem. C 112, 1305 (2008).
[12] W. J. Mitchell et al., "Hydrogenation of Carbon Monoxide at 100 K on the Ruthenium(001) Surface: Spectroscopic Identification of Formyl Intermediates," J. Am. Chem. Soc. 115, 4381 (1993).