| h1 = s + px + py + pz | h2 = s + px - py - pz | h3 = s - px + py - pz | h4 = s - px - py + pz |
On atom #2 we have
| h5 = s - px - py - pz | h6 = s - px + py + pz | h7 = s + px - py + pz | h8 = s + px + py - pz |
We do this because the energy gap is caused by the bonding interaction V between hybrids pointing at each other from adjacent sites. Were this the only interaction in the problem, the two hybrids would mix and spit into bonding and antibonding states separated by energy 2V. These, in turn, would become degenerate valence and conduction bands, which would be flat and degnerate. However, V isn't the only interaction in the problem. The remaining ones broaden out the bands in complex ways. You can model the band structures of all the materials in this class by adjusting the interactions.
Let us now assume that the only interaction other than V is the energy splitting between the s and p orbitals on a given site. We neglected this splitting when we combined them into hybrids. This assumption is oversimplified for Si and Ge, so the bands that result don't match these materials well. However, the qualitative features are right. In the hybrid basis, the s-p splitting becomes a matrix element Vh to tunnel from any hybrid orbital to any other on the same site. With this choice of interaction, the 8 x 8 matrix that we must diagonalize for every crystal momentum q becomes
| 0 | Vh | Vh | Vh | V eiq·&alpha | 0 | 0 | 0 | |
| Vh | 0 | Vh | Vh | 0 | V eiq·&beta | 0 | 0 | |
| Vh | Vh | 0 | Vh | 0 | 0 | V eiq·&gamma | 0 | |
| Vh | Vh | Vh | 0 | 0 | 0 | 0 | V eiq·&delta | |
| V e-iq·&alpha | 0 | 0 | 0 | 0 | Vh | Vh | Vh | |
| 0 | V e-iq·&beta | 0 | 0 | Vh | 0 | Vh | Vh | |
| 0 | 0 | V e-iq·&gamma | 0 | Vh | Vh | 0 | Vh | |
| 0 | 0 | 0 | V e-iq·&delta | Vh | Vh | Vh | 0 |
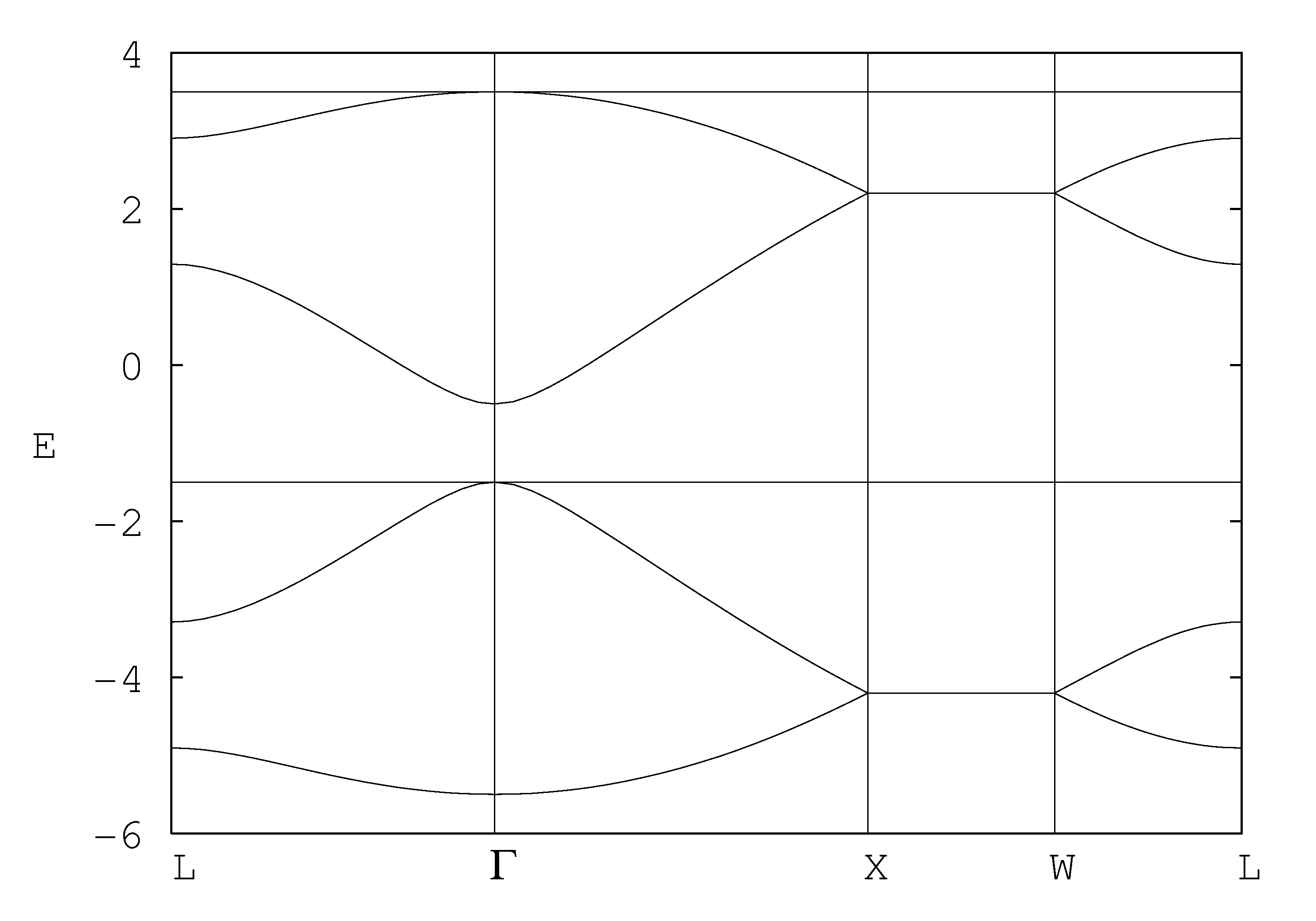
While this particular matrix is sufficiently simple that you can obtain some of its eigenvalues by hand, actually doing so is not useful because the mathematical simplicity is an artifact of the parameter choices. It makes more sense to write a computer program that can diagonalize any matrix, and then save the code for later modification to study other sets of parameters. The bands shown in Fig. 1 were generated by such a code, the fortran source of which is available here. The figure itself was generated by gnuplot, the input file of which is available here.
The band structure in Fig. 1 is a sequence of straight-line trajectories between particular values of q, given by
| Γ= (0 , 0, 0) | L= π√3/4 b (1 , 1 , 1) | X= π√3/2 b (1 , 0 , 0) | W= π√3/4 b (2 , 1 , 0) |
where b = |α| = ... = |&delta| is the bond length. It consists of four valence bands (E < -1) and four conductions bands (E > -1). The total number of bands is thus the number of orbitals in the unit cell, as expected. The double degeneracy of the flat bands (and also the fact that they are flat) is an artifact of the model that disappears when other interactions are included.
© 2007 R. B. Laughlin. The author grants permission to copy, distribute and display this work in unaltered form, with attribution to the author, for noncommercial purposes only. All other rights, including commercial rights, are reserved to the author.
References
[1] W. Harrison, Elementary Electronic Structure (World, Singapore, 2004).